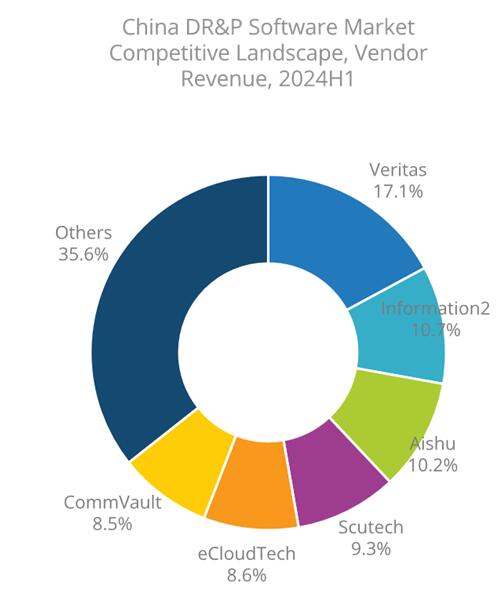
Summit超级计算机对10,000原子镁错位系统进行了仿真,可以让科学家了解哪些合金材料添加到提高镁的延展性。
密歇根大学机械工程副教授Vikram Gavini领导的团队使用了橡树岭实验室的Summit超级计算机对10,000原子镁错位系统进行了仿真,这一壮举可以让科学家了解哪些合金材料添加到提高镁的延展性。通过模拟比以前的代码大10倍,快20倍的系统,该团队最终实现了200倍的提速,以46 petaflops的速度运行,并获得了美国计算机协会的戈登·贝尔奖入围奖。

美国summit超级计算机
加维尼说:“使用Summit这台世界排名第一的超级计算机,我们能够对镁错位系统进行迄今为止最大的模拟,我们认为这对提高其延展性至关重要。” 如果科学家可以高精度模拟合金,他们将能够在实验测试之前对新材料进行预筛分,从而加快了新材料的引入。借助对某些类型的位错所涉及的能量原理的了解,实验者可以更轻松地为新合金提出建议。
为了理解材料,Gavini的团队执行了密度泛函理论(DFT)计算,该计算专注于材料系统的电子结构。到目前为止,现有的DFT代码还无法处理准确建模这些缺陷所需的大型系统。为了解决这些局限性,多年来,Gavini的研究小组已经开发出了用于快速,准确和可扩展DFT计算的方法和算法。
去年,团队成员参加了OLCF黑客马拉松,这是一次动手实践的研讨会,研究人员在专家指导下将他们现有的代码移植到GPU。在黑客马拉松之前,他们没有现有的GPU代码,但是在活动中,他们能够将其代码转移到Summit上,Summit是世界上功能最强大,最智能的开放科学超级计算机。
“我们所有的算法在CPU上都是稳定的,但是我们知道我们必须有一个基于GPU的代码才能充分利用Summit之类的架构,” Gavini在美国能源部 (DOE) OLCF上谈到该系统时说道。在通过Director的全权分配获得在Summit上的时间之后,团队花了6个月的时间将其90%的代码DFT-FE(带有有限元素的DFT)移植到GPU。研究人员在进行最后的10,000原子模拟之前,在Summit上对镁的位错系统进行了较小的模拟,其中包含约2,000个原子。最终的模拟结果为13.6纳米乘以13.6纳米乘以1.2纳米,为团队提供了了解镁系统高保真的能量特性所需的精度。
研究人员希望该代码的GPU加速版本将成为科学家预先筛选和确定实验者可以进一步改进的关键合金的计算工具。
声明: 此文观点不代表本站立场;转载须要保留原文链接;版权疑问请联系我们。